ou’re not just blocking an allergy response. You’re suppressing one of the most fundamental neurotransmitters your body runs on.
Millions of people take antihistamines daily — for allergies, for sleep, for mast cell activation, for hives that won’t quit. And most of them have no idea that these drugs reach far beyond histamine. They’re quietly interfering with acetylcholine, the neurotransmitter that controls your gut motility, your memory, your vagus nerve tone, and your body’s master anti-inflammatory switch.
This isn’t a fringe concern. This is a mechanistic reality that the research has made very clear — and yet almost nobody talks about it.
Let me walk you through exactly what’s happening.
What Is the Cholinergic System?
The cholinergic system is the network of neurons and receptors that use acetylcholine (ACh) as their signaling molecule. Acetylcholine was the first neurotransmitter ever identified, and for good reason — it’s everywhere.
ACh is synthesized from two precursors: acetyl-CoA (a mitochondrial metabolite) and choline (a dietary nutrient). The enzyme choline acetyltransferase (ChAT) combines these to produce acetylcholine, which is then packaged into vesicles and released at nerve terminals.
Here’s what acetylcholine actually does in your body:
In the brain: ACh is critical for memory formation, learning, attention, and executive function. It drives the encoding of new memories in the hippocampus and maintains cognitive sharpness through cortical activation. This is why Alzheimer’s disease — the quintessential disease of memory loss — is fundamentally a disease of cholinergic neuron death.
In the gut: ACh is the dominant neurotransmitter of the enteric nervous system — your “second brain.” It directly controls intestinal contractions, coordinates the migrating motor complex (the cleansing wave that sweeps between meals), regulates mucus secretion from goblet cells, controls chloride secretion for mucosal hydration, and modulates epithelial barrier integrity. Without adequate cholinergic signaling, motility stalls, secretions dry up, and the mucosal surface becomes vulnerable.
In the autonomic nervous system: ACh is the neurotransmitter of the entire parasympathetic nervous system — the “rest and digest” arm. It controls heart rate deceleration, bronchial secretion, salivation, tear production, urinary bladder contraction, and pupil constriction. Every time your body shifts from sympathetic overdrive into recovery mode, that transition is mediated by acetylcholine.
At the neuromuscular junction: ACh is the sole neurotransmitter responsible for every voluntary muscle contraction in your body. Motor neurons release ACh onto nicotinic receptors at the motor end plate, triggering the depolarization that produces muscle fiber contraction. Whether you’re gripping, lifting, walking, or breathing — it all runs through acetylcholine.
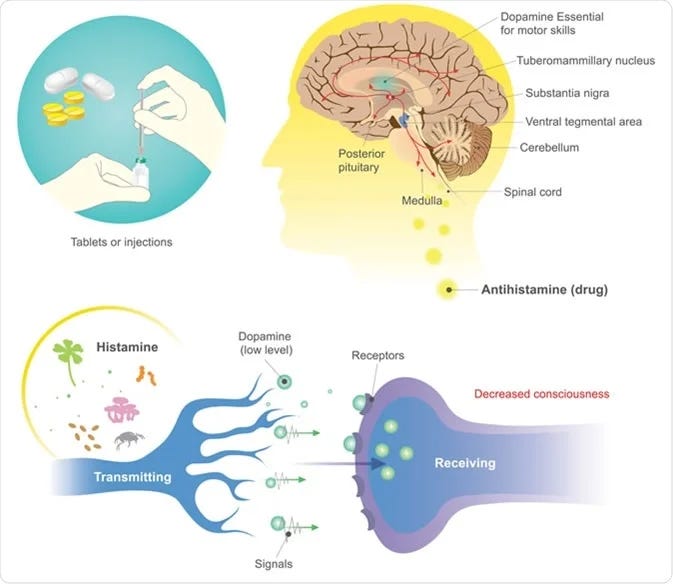
In dopamine regulation: This is a connection almost nobody makes. Cholinergic neurons from the mesopontine nuclei send dense projections to dopamine neurons in the ventral tegmental area (VTA), and cholinergic interneurons in the striatum directly trigger local dopamine release. Acetylcholine is a primary regulator of mesolimbic dopamine output — the circuit that controls motivation, drive, reward processing, and goal-directed behavior. Suppress acetylcholine, and dopamine transmission drops with it.
In the immune system: This is where it gets profound. ACh is the molecular effector of the cholinergic anti-inflammatory pathway (CAP) — a vagus nerve–dependent circuit that directly suppresses inflammatory cytokine production from macrophages and, critically, mucosal mast cells. The vagus nerve releases ACh, which binds to α7 nicotinic acetylcholine receptors (α7nAChR) on immune cell surfaces, inhibiting NF-κB signaling and suppressing TNF-α, IL-1β, IL-6, and IL-18 at the post-transcriptional level. This pathway was first described by Kevin Tracey’s group at the Feinstein Institutes and published in Nature (Wang et al., 2003). It fundamentally reshaped our understanding of neuro-immune communication.
When you understand this, you realize that acetylcholine isn’t just “a” neurotransmitter. It’s the signaling backbone of parasympathetic function, gut motility, cognitive processing, and inflammatory regulation. It is, by any measure, one of the most important molecules in human physiology.
ow Antihistamines Work — And Why the Problem Starts
Antihistamines are classified as H1 receptor inverse agonists. Technically, they don’t just “block” the receptor — they stabilize the histamine H1 receptor in its inactive conformation, pushing the equilibrium away from constitutive signaling. This is why the more precise pharmacological term is “inverse agonist” rather than “antagonist.”
There are two generations:
First-generation antihistamines — diphenhydramine (Benadryl), promethazine (Phenergan), chlorpheniramine, hydroxyzine, cyproheptadine — were developed in the 1940s. Here’s the critical fact: these drugs were born from the same chemical scaffold used to create muscarinic cholinergic antagonists, tranquilizers, and antipsychotics. They share approximately 45% sequence homology between H1 and muscarinic acetylcholine receptor binding domains. This isn’t an accident — it’s a structural inevitability.
These first-generation drugs cross the blood-brain barrier freely, bind muscarinic receptors in the CNS, and produce the full spectrum of anticholinergic effects: sedation, cognitive impairment, dry mouth, constipation, urinary retention, blurred vision, and tachycardia.
Second-generation antihistamines — loratadine (Claritin), cetirizine (Zyrtec), fexofenadine (Allegra), desloratadine (Clarinex) — were designed to be more selective for H1 receptors and to minimize blood-brain barrier penetration. But here’s what most people don’t know: several of these drugs still possess measurable anticholinergic activity.
Research published in the European Journal of Pharmacology (Orzechowski et al., 2005) ranked the anticholinergic potencies of 10 antihistamines in two functional models. The rank order was: cyproheptadine > promethazine > desloratadine > diphenhydramine > loratadine > chlorpheniramine > hydroxyzine > pyrilamine. Notably, fexofenadine and cetirizine showed no measurable anticholinergic activity in either model — making them the cleanest options from a cholinergic standpoint.
Desloratadine and loratadine — both widely marketed as “non-sedating” and therefore presumed safe — showed clear anticholinergic effects in both in vitro and in vivo models. This means even some second-generation antihistamines are quietly suppressing acetylcholine signaling.
What Happens When You Block Acetylcholine
When antihistamines bind muscarinic receptors and suppress cholinergic transmission, the downstream consequences hit multiple systems simultaneously:
Cognitive Function
Acetylcholine drives memory encoding, sustained attention, and working memory through M1 muscarinic receptor activation in the cortex and hippocampus. Block this, and you get the classic anticholinergic cognitive profile: brain fog, difficulty forming new memories, slowed processing speed, and impaired concentration.
First-generation antihistamines taken at bedtime don’t just cause drowsiness — they disrupt REM sleep architecture, increasing latency to REM onset and reducing total REM duration. The residual cognitive impairment — impaired attention, working memory deficits, and slowed reaction time — persists into the next morning, even after the subjective sedation has resolved.
The long-term implications are even more concerning. A meta-analysis published in Age and Ageing (Pieper et al., 2020) pooled data from 26 studies encompassing over 621,000 participants and found that use of strongly anticholinergic drugs was associated with a 1.20-fold increased risk of incident dementia, with long-term use (365+ days) elevating risk to 1.50-fold. A landmark nested case-control study from JAMA Internal Medicine (Coupland et al., 2019) involving over 284,000 participants found significant dose-response relationships, with the population-attributable fraction estimated at approximately 10% — meaning roughly 1 in 10 dementia cases in the study population could theoretically be attributed to anticholinergic drug exposure.
The biological plausibility is straightforward: older adults already experience age-related decline in acetylcholine synthesis and receptor density. Layering anticholinergic medications on top of this age-related cholinergic deficit creates a compounding vulnerability. The American Geriatrics Society Beers Criteria now explicitly classifies first-generation antihistamines as potentially inappropriate medications in adults over 65.
Acetylcholine Drives Dopamine Release — And That’s Why You Feel Flat
Here’s something that almost nobody connects to their antihistamine use: acetylcholine is a primary regulator of dopamine release in the brain.
The mesolimbic dopamine pathway — the circuit responsible for motivation, reward processing, drive, and goal-directed behavior — originates in the ventral tegmental area (VTA) of the midbrain and projects to the nucleus accumbens. This circuit doesn’t operate in isolation. It is governed by cholinergic input from the mesopontine nuclei — specifically the pedunculopontine and laterodorsal tegmental nuclei — which send dense cholinergic projections directly to VTA dopamine neurons.
When acetylcholine binds to nicotinic receptors (primarily α4β2 and α6β2 subtypes)* on these dopamine neurons, it directly modulates their firing rate and pattern, including the transition from tonic to phasic burst firing — which is the precise firing mode that encodes reward, salience, and motivational drive. Research published in Nature Neuroscience (Zhou et al., 2001) demonstrated that in striatal slices, blocking nicotinic receptors or depleting endogenous acetylcholine reduced evoked dopamine release by up to 90%. That is not a marginal modulation. That is acetylcholine functioning as a gatekeeper of dopamine transmission.
Additionally, cholinergic interneurons within the striatum itself directly trigger local dopamine release from dopaminergic terminals, independent of VTA cell body firing. Research from Bhatt and colleagues (Threlfell et al., Neuron, 2012; Cachope et al., Cell Reports, 2012) showed that synchronized activity of these cholinergic interneurons is sufficient to drive phasic dopamine release in the nucleus accumbens. Acetylcholine is not just modulating dopamine from upstream — it’s triggering it locally at the terminal level.
This is why people on chronic anticholinergic antihistamines report low motivation, emotional flatness, anhedonia, and difficulty initiating tasks. These aren’t vague “side effects.” They are the predictable neurochemical consequence of suppressing cholinergic tone in a circuit where acetylcholine directly controls dopamine output. You cannot block acetylcholine and expect your motivational drive to stay intact. The wiring doesn’t allow it.
Skeletal Muscle and the Neuromuscular Junction
Acetylcholine isn’t just a brain and gut neurotransmitter — it is the neurotransmitter at every neuromuscular junction in your body. Every voluntary muscle contraction you initiate — whether you’re gripping a barbell, climbing stairs, or simply standing upright — depends on motor neurons releasing ACh onto nicotinic receptors at the motor end plate.
When ACh binds nicotinic receptors on skeletal muscle fibers, it triggers the depolarization cascade that produces muscle contraction. This is a non-negotiable step in voluntary movement. Anticholinergic drugs that reach the periphery can interfere with this transmission, contributing to muscle weakness, reduced exercise performance, slowed reaction time, and impaired coordination. Research has confirmed that antihistamines with anticholinergic properties — even some second-generation agents like loratadine and desloratadine — can produce measurable deficits in reaction time and increase physiological tremor, effects attributable to disrupted cholinergic neurotransmission affecting motor control (Baumann-Birkbeck et al., Clinical Neurophysiology, 2014).
For anyone training in the gym, this matters. Your ability to recruit muscle fibers, maintain grip strength, and sustain neuromuscular efficiency during a session is fundamentally dependent on intact cholinergic signaling at the neuromuscular junction. Suppressing this system pharmacologically is working directly against your training adaptations.
Gut Motility, the Migrating Motor Complex, and Chronic Gut Dysfunction
Acetylcholine is the primary excitatory neurotransmitter of the enteric nervous system. It directly drives peristaltic contractions through M2 and M3 muscarinic receptor activation on intestinal smooth muscle. M3 receptors generate the primary contractile response, while M2 receptors prevent relaxation by inhibiting adenylyl cyclase.
ACh also regulates the interstitial cells of Cajal (ICCs) — the pacemaker cells of the gut. These cells generate the slow-wave electrical activity that coordinates the rhythmic contractions of the intestine. Cholinergic input modulates ICC pacemaker frequency and propagation patterns. Disrupting this input doesn’t just slow motility — it disorganizes the fundamental electrical coordination of gut movement.
Critically, the migrating motor complex (MMC) — the powerful cyclic “cleansing wave” that sweeps undigested material, bacteria, and debris through the small intestine between meals — is highly regulated by both serotonin and acetylcholine. The MMC depends on coordinated activation of cholinergic motor neurons within the myenteric plexus. Serotonin (5-HT), released from enterochromaffin cells, activates 5-HT4 receptors on enteric neurons, which in turn triggers acetylcholine release from cholinergic motor neurons to drive the contractile phase of the MMC. This is a serotonin→acetylcholine relay — and both neurotransmitters must be functioning for the MMC to operate properly. When you pharmacologically reduce cholinergic tone with anticholinergic antihistamines, you are disrupting the effector arm of this relay, even if serotonin signaling is intact. The MMC loses its contractile power, and bacterial overgrowth risk increases because the small intestine is no longer being adequately cleared.
Beyond motility, ACh regulates mucus secretion from goblet cells, chloride secretion that maintains mucosal hydration, epithelial proliferation, and barrier function. Colonic epithelial cells themselves produce non-neuronal acetylcholine through ChAT expression, and this epithelial ACh participates in the secretory response to short-chain fatty acids like propionate and butyrate.
This is why anticholinergic side effects in the gut aren’t just “constipation” — they represent a broad suppression of mucosal defense, secretory function, MMC cycling, and motility coordination. The cumulative result is what I call chronic gut dysfunction: the progressive breakdown of the coordinated neuromuscular and secretory programs that keep the gut functional. For anyone already dealing with compromised gut function — SIBO, motility disorders, or mucosal barrier failure — adding anticholinergic load is not just unhelpful. It is mechanistically driving the very dysfunction you’re trying to resolve.
The α7 Nicotinic Acetylcholine Receptor — The Most Underrated Anti-Inflammatory Target in Your Body
This is where the story becomes especially relevant for anyone dealing with chronic inflammation, mast cell activation, or autoimmune conditions. And this is the piece that almost everyone — patients and practitioners alike — is missing.
The α7 nicotinic acetylcholine receptor (α7nAChR) is a ligand-gated ion channel that forms a homopentameric structure made entirely of α7 subunits. Its endogenous ligand is acetylcholine. And it is, without exaggeration, one of the most important anti-inflammatory receptors in the human body.
The α7nAChR is the molecular effector of the cholinergic anti-inflammatory pathway (CAP) — a neural circuit in which the efferent vagus nerve releases acetylcholine that acts on α7nAChRs expressed on macrophages, dendritic cells, and critically, mucosal mast cells. When activated, this receptor triggers JAK2-STAT3 signaling and inhibits NF-κB nuclear translocation, resulting in potent suppression of pro-inflammatory cytokine release — specifically TNF-α, IL-1β, IL-6, and IL-18 — without affecting anti-inflammatory IL-10.
This is not a subtle effect. In animal models, vagus nerve stimulation dramatically reduces systemic inflammatory responses, improves survival in sepsis models, and attenuates tissue damage in ischemia-reperfusion injury, arthritis, inflammatory bowel disease, and pancreatitis. The α7nAChR has been confirmed as the essential receptor mediating these effects — vagus nerve stimulation fails to suppress TNF in α7-deficient mice (Wang et al., Nature, 2003).
Where α7nAChR is expressed tells you everything about why it matters:
In the gut lining: Functional α7nAChRs are expressed on intestinal epithelial cells, submucosal plexus neurons, and resident immune cells throughout the gut wall. The receptor is present in both the small intestine and colon, where it regulates epithelial barrier integrity, mucus secretion, immune cell activation within the lamina propria, and the inflammatory tone of the mucosal environment. It is positioned at the exact interface where the immune system meets the microbial ecosystem — making it a frontline regulator of intestinal immune homeostasis.
On mast cells: This is the connection that should stop every MCAS patient in their tracks. Research has demonstrated that mucosal mast cells express α7nAChRs, and that activation of these receptors directly inhibits antigen-IgE-mediated mast cell degranulation. In a murine food allergy model (Hori et al., PLOS ONE, 2014), treatment with the selective α7nAChR agonist GTS-21 and vagal stimulation with 2-deoxy-D-glucose both significantly alleviated allergic symptoms by suppressing mucosal mast cell activation through α7nAChR signaling. The mast cells in this model were localized primarily in the mucosal epithelium — the exact position where they orchestrate allergic and inflammatory responses. Additionally, nicotine has been shown to inhibit Fcε receptor-induced cysteinyl leukotriene and cytokine production from mast cells through α7/α9/α10 nicotinic receptor pathways without blocking degranulation itself (Mishra et al., Journal of Immunology, 2010). This means the α7nAChR modulates the inflammatory output of mast cells — the cytokine storm — not just granule release. For anyone with mast cell activation syndrome, the α7nAChR is arguably the single most important endogenous brake on mast cell–driven inflammation.
In the brain — neuroprotection: The α7nAChR is highly expressed
in brain regions critical for memory and cognition, including the hippocampus and prefrontal cortex. It is implicated in long-term memory formation, sensorimotor gating, and neuroprotection against inflammatory neurotoxicity. Preclinical research has demonstrated protective roles for α7nAChR signaling in models of stroke, Alzheimer’s disease, and neuroinflammation. The receptor reduces neuroinflammatory damage by suppressing microglial activation through the same NF-κB inhibition pathway it uses in peripheral macrophages. Both α4β2 and α7 nicotinic receptors are critical for memory, working memory, learning, and attention — and α7 agonists have shown positive effects on neurocognition in clinical populations including individuals with schizophrenia. When anticholinergic medications suppress the ligand that activates this receptor, you are stripping away a fundamental layer of neuroprotection.
The MCAS paradox — stated plainly: Drugs that reduce cholinergic tone — including antihistamines with anticholinergic properties — are working against this protective circuit. You cannot simultaneously suppress acetylcholine signaling and expect the vagal anti-inflammatory reflex to function normally. For someone using antihistamines to manage mast cell–driven inflammation, the pharmacological irony is devastating: you may be reducing histamine at the H1 receptor level while simultaneously disabling the α7nAChR-mediated brake on mast cell inflammatory output. You are treating one output of the mast cell while removing the body’s own mechanism for controlling the cell itself. This is not a theoretical concern — this is the mechanistic reality of what anticholinergic antihistamines do to the cholinergic anti-inflammatory pathway.
Autonomic Balance
Every symptom that anticholinergic drugs produce — dry mouth, constipation, urinary retention, tachycardia, reduced sweating, blurred vision — represents a specific loss of parasympathetic function. What this amounts to in aggregate is a pharmacological shift toward sympathetic dominance.
For anyone with existing dysautonomia, POTS, or autonomic dysfunction, this shift is the opposite of therapeutic. These individuals already have impaired parasympathetic tone. Adding anticholinergic load deepens the imbalance.
What You Need to Support If You’re Taking Antihistamines
If antihistamines are clinically necessary for you — and for many people with MCAS, severe allergies, or chronic urticaria, they are — here’s what the mechanism tells us about supporting the systems being suppressed:
Choline intake. Choline is the rate-limiting substrate for acetylcholine synthesis. Dietary sources include eggs (particularly yolks), liver, wild-caught fish, and cruciferous vegetables. The adequate intake for adults is 550 mg/day for men and 425 mg/day for women, though many people fall well short. Supplemental forms include CDP-choline (citicoline) and alpha-GPC, both of which cross the blood-brain barrier and support central cholinergic function.
Mitochondrial support. Acetylcholine synthesis requires acetyl-CoA, which is a direct output of mitochondrial metabolism. Anything that supports mitochondrial bioenergetics — adequate B vitamins (B1, B5 in particular), CoQ10, magnesium, adequate caloric intake from whole foods — indirectly supports the substrate pool for ACh production.
Vagal tone practices. Since the cholinergic anti-inflammatory pathway depends on vagus nerve output, practices that enhance vagal tone become mechanistically relevant: slow diaphragmatic breathing, cold water face immersion (the dive reflex), gargling, singing, and regular aerobic exercise all enhance vagal efferent activity.
Choose your antihistamine carefully. If you need H1 blockade, fexofenadine has the lowest anticholinergic activity among commonly used antihistamines — essentially zero in functional models. Cetirizine also shows negligible anticholinergic effects, though it does cause more sedation than fexofenadine. If you’re using diphenhydramine, hydroxyzine, or promethazine regularly, you’re carrying a significant anticholinergic burden.
Assess total anticholinergic burden. Antihistamines are rarely the only anticholinergic drug someone is taking. Antidepressants (especially tricyclics and paroxetine), bladder medications (oxybutynin), muscle relaxants (cyclobenzaprine), and some antipsychotics all carry anticholinergic activity. The cumulative burden — called the Anticholinergic Burden (ACB) score — is what drives risk, not any single drug in isolation.
The Bottom Line
Acetylcholine is not optional. It is the neurotransmitter of parasympathetic recovery, gut motility, the migrating motor complex, cognitive function, dopamine-driven motivation, neuromuscular performance, and — through the α7 nicotinic receptor — your most powerful endogenous brake on mast cell and macrophage-driven inflammation. Every antihistamine with anticholinergic properties is — to varying degrees — suppressing this entire system.
This doesn’t mean antihistamines are never appropriate. It means the decision to use them should be made with full mechanistic awareness of what you’re trading. And it means that if you must use them, you should actively support the cholinergic system through substrate provision, vagal tone practices, and careful drug selection.
The cholinergic system is not background noise. It is the operating system. And the α7nAChR — expressed on your mast cells, your gut epithelium, your macrophages, and your neurons — is one of the most underappreciated therapeutic targets in medicine. Treat it accordingly.
References
Wang, H., Yu, M., Ochani, M., et al. (2003). Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature, 421, 384–388.
Tracey, K. J. (2002). The inflammatory reflex. Nature, 420, 853–859.
Pavlov, V. A., Wang, H., Czura, C. J., et al. (2003). The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Molecular Medicine, 9(5–8), 125–134.
Orzechowski, R. F., Currie, D. S., & Valancius, C. A. (2005). Comparative anticholinergic activities of 10 histamine H1 receptor antagonists in two functional models. European Journal of Pharmacology, 506(3), 257–264.
Pieper, N. T., Grossi, C. M., Chan, W. Y., et al. (2020). Anticholinergic drugs and incident dementia, mild cognitive impairment and cognitive decline: a meta-analysis. Age and Ageing, 49(6), 939–947.
Coupland, C. A. C., Hill, T., Dening, T., et al. (2019). Anticholinergic drug exposure and the risk of dementia: a nested case-control study. JAMA Internal Medicine, 179(
Gray, S. L., Anderson, M. L., Dublin, S., et al. (2015). Cumulative use of strong anticholinergics and incident dementia: a prospective cohort study. JAMA Internal Medicine, 175(3), 401–407.
Yajima, T., et al. (2011). Non-neuronal release of ACh plays a key role in secretory response to luminal propionate in rat colon. Journal of Physiology, 589(4), 953–962.
Harrington, A. M., Hutson, J. M., & Southwell, B. R. (2010). Cholinergic neurotransmission and muscarinic receptors in the enteric nervous system. Progress in Histochemistry and Cytochemistry, 44(4), 173–202.
Rosas-Ballina, M., et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science, 334(6052), 98–101.
Church, M. K., & Church, D. S. (2013). Pharmacology of antihistamines. Indian Journal of Dermatology, 58(3), 219–224.
Keever, K. R., et al. (2024). Cholinergic signaling via the α7 nicotinic acetylcholine receptor regulates the migration of monocyte-derived macrophages during acute inflammation. Journal of Neuroinflammation, 21, 3.
Hori, M., & Bhatt, D. (2023). Role of muscarinic acetylcholine receptors in intestinal epithelial homeostasis. International Journal of Molecular Sciences, 24(7), 6508.
Zhou, F. M., Liang, Y., & Dani, J. A. (2001). Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nature Neuroscience, 4(12), 1224–1229.
Threlfell, S., Lalic, T., Platt, N. J., et al. (2012). Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron, 75(1), 58–64.
Cachope, R., Mateo, Y., Mathur, B. N., et al. (2012). Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Reports, 2(1), 33–41.
Xie, Y., et al. (2019). Neural circuits and nicotinic acetylcholine receptors mediate the cholinergic regulation of midbrain dopaminergic neurons and nicotine dependence. Acta Pharmacologica Sinica, 40, 1–10.
Hori, M., et al. (2014). Anti-allergic role of cholinergic neuronal pathway via α7 nicotinic ACh receptors on mucosal mast cells in a murine food allergy model. PLOS ONE, 9(1), e85886.
Mishra, N. C., et al. (2010). Nicotine inhibits FcεRI-induced cysteinyl leukotrienes and cytokine production without affecting mast cell degranulation through α7/α9/α10-nicotinic receptors. Journal of Immunology, 185(1), 588–596.
de Jonge, W. J., & Bhatt, D. (2007). The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. British Journal of Pharmacology, 151, 915–929.
Báez-Pagán, C. A., Delgado-Vélez, M., & Bhatt, D. (2015). Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. Journal of Neuroimmune Pharmacology, 10, 468–476.
Baumann-Birkbeck, L., et al. (2014). The effects of antihistamines with varying anticholinergic properties on voluntary and involuntary movement. Clinical Neurophysiology, 125(